The
metabolism, distribution and elimination of medications can occur through a
number of pathways which can involve the cytochrome P450 (CYP) enzyme system
(phase I metabolism), conjugative enzymes such as sulfation and
UDP-glucuronosyltransferase (UGT; phase II metabolism), and influx/efflux cell
membrane transporters.(1-3) In addition, it is now known that many
medications can be influenced by a combination of these pathways, thus
influencing their safety and efficacy profiles as well as the pharmacokinetic
profiles of other medications. To complicate things further, all of these
pathways are known to be subject to genetic polymorphisms, which can contribute
to the differences in the pharmacokinetic and pharmacodynamic properties of a
particular medication. It is important for the clinician to recognize and
appreciate the contribution of these pathways for proper implementation of drug
therapy into clinical practice.
As
it relates to the metabolism of a medication, the CYP450 enzyme system
contributes the most, with CYP3A4 being the most common enzyme used in the
metabolism of many medications.(1-3) The second largest contributor to drug
metabolism occurs through UGT enzymes.(3) There are currently eleven
different UGT enzymes that are known to contribute to the phase II metabolism
of many of the most prescribed medications used in clinical practice.
Fortunately, the number and degree of significance of drug-drug interactions
associated through the UGT enzymes are not as problematic when compared to
those that rely upon the CYP450 enzyme system.3 Of the eleven UGT
enzymes, UGT2B7 appears to contribute to the metabolism of the greatest number
of medications. The presence of UGT2B7 is similar to locations for other
enzymes of drug metabolism and includes the intestine, liver and kidney.
Following UGT2B7 in the number medications that are substrates, are UGT1A1,
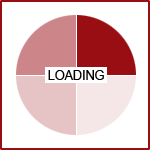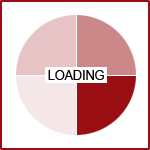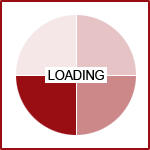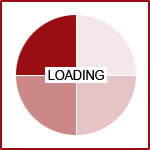
UGT1A9, and UGT1A4. UGT2B7 is used by up to 35% of medications on the market, followed by UGT1A4 which was
used by 20% and UGT1A1 by 15%.(3)
Therefore,
regardless of the perspective of the evaluation of the UGT enzyme system on
drug metabolism, UGT2B7 remains to be one of the greatest contributors of drug
metabolism for medications that clinicians will likely prescribe, dispense
and/or administer to their patients. In fact, it is the inhibition of
UGT2B7 by valproic acid that contributes to the life threatening skin rashes
when being coadministered with the anticonvulsant, lamotrigine (Lamictal).4
Since lamotrigine does not have many other metabolic pathways to go through,
this drug-drug interaction with valproic acid can significantly compromise the
safety of the patient. Another example of a clinically relevant drug
interaction is known to occur with UGT1A1, where known inhibitors of UGT1A1
(such as atazanavir, gemfibrozil, and indinavir) could put the patient taking
irinotecan at significant risk for bone marrow suppression since irinotecan is
largely dependent on UGT1A1 for its metabolism.(5-7)
Since
many medications have several other UGT enzymes for their metabolism, as well
the use of CYP450 enzymes and/or transporters, the inhibition of one UGT enzyme
alone generally does not result in clinically relevant drug interactions for
some medications.(3) This does not mean that drug interactions
or adverse drug events are not associated with UGT enzymes. The above
examples clearly show that their influence can be clinically significant.